Poster un message
En réponse à :
Effets tissulaires des UV
I. Erythème et cellules « coup de soleil »
L’un des premiers effets tissulaires de l’exposition UV est l’érythème (« coup de soleil »), une réponse inflammatoire provoquant la vasodilatation des vaisseaux sanguins et le rougissement de la peau. Cette réaction est visible approximativement 6 à 8 heures après l’exposition UV et disparaît au bout de 36 à 48 heures. Les facteurs à l’origine de l’érythème sont encore mal connus, mais la présence de dimères de pyrimidines dans l’ADN pourrait déclencher cette réponse inflammatoire car il a été montré que le spectre d’activité de l’érythème est très proche de celui de la formation des CPD (Young et al., 1998). La dose minimale érythémateuse (DEM ) définit la dose à partir de laquelle l’érythème cutané est observé in vivo. La DEM dépend du phototype cutané, défini par les caractéristiques physiques de l’individu (couleur de la peau, des yeux, des cheveux) et par la réaction de leur peau au soleil (tableau ci-dessous).
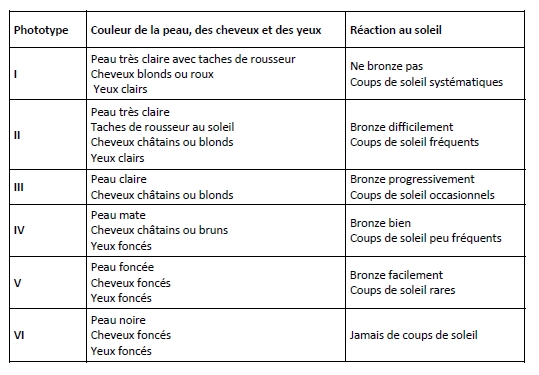
- Phototypes cutanés
Parallèlement à la réaction érythémateuse, l’exposition de la peau aux UV provoque l’apparition de kératinocytes apoptotiques appelés « sunburn cells » (SBC, cellules « coup de soleil »). Ils sont caractérisés par un noyau pycnotique (rétraction du noyau sous l’effet de la condensation de la chromatine) et un cytoplasme éosinophile vacuolisé (Brash et al., 1996). La formation des SBC est dépendante de p53 (Ziegler et al., 1994) et constituerait un mécanisme de protection permettant l’élimination des cellules présentant un risque de transformation maligne (e.g. excès de lésions). La dose biologique efficace (DEB) définit la dose UV minimale capable d’induire la formation de SBC dans les couches suprabasales de l’épiderme 24h après l’exposition. La DEB est utilisée pour les études ex vivo (peaux reconstruites, biopsies irradiées ex vivo) quand la DEM ne peut être définie (absence d’érythème). Il existe une étroite corrélation entre la DEB et la DEM (Bernerd and Asselineau, 1997).
II. Pigmentation induite par les UV
La mélanine , répartie en bouclier au dessus du noyau des kératinocytes de l’épiderme, joue un rôle protecteur contre les rayonnements UV : elle agit à la fois comme une barrière physique qui disperse les rayons UV et comme un filtre qui absorbe une partie des rayonnements, limitant ainsi leur pénétration dans la peau (Brenner and Hearing, 2008). La pigmentation constitutive de la peau détermine la sensibilité de celle‐ci à l’exposition UV. Ainsi les peaux noires, plus riches en eumélanine que les peaux claires, sont mieux protégées contre les dommages induits par les UV (Del Bino et al., 2006 ; Yamaguchi et al., 2006).
Le mécanisme d’induction de la pigmentation après une exposition UV dépend de la longueur d’onde du rayonnement. La coloration immédiate et persistante de la peau observée après une irradiation UVA serait liée à l’oxydation de la mélanine pré‐existante, puis de ses précurseurs, et à leur redistribution vers les dendrites périphériques des mélanocytes , provoquant une accumulation de pigments bruns dans les différentes couches de l’épiderme (Maeda and Hatao, 2004 ; Routaboul et al., 1999). Les UVB stimulent la production de la mélanine en induisant l’activité de la tyrosinase, une enzyme localisée dans les mélanosomes . Cette activité s’accompagne d’une augmentation du nombre et de la dendricité des mélanocytes, favorisant le transfert d’un nombre croissant de mélanosomes vers les kératinocytes (Brenner and Hearing, 2008). Ce processus permet en quelques jours la mise en place d’une photo‐protection durable (Gilchrest et al., 1996).
III. Immunosuppression induite par les UV
Voir Immunosuppression induite par les UV par M Démarchez
IV. Photovieillissement
IV.1. Caractéristiques cliniques et biochimiques du photovieillissement
Le photovieillissement, ou vieillissement actinique, est un phénomène qui résulte des expositions chroniques de la peau au soleil. Il est à dissocier du vieillissement chronologique , ou intrinsèque, qui intervient naturellement au cours du temps. D’un point de vue clinique, la peau photoâgée se caractérise par un aspect papyracé reflétant sa fragilité, sa perte d’élasticité et sa sécheresse, et par la présence de rides profondes et de zones d’hyper‐ ou d’hypopigmentation. La présence de kératoses actiniques, les lésions précurseurs des SCC, est également fréquente (Berneburg et al., 2000).
Au niveau épidermique, les modifications liées au photovieillissement sont minimes et diffèrent peu de celles associées au vieillissement chronologique. L’épiderme est aminci et la jonction épidermique présente un aplatissement et une perte des crêtes épidermiques. Au contraire, des altérations majeures sont observées dans le derme , avec des modifications très importantes de la structure et de l’organisation de la MEC. La caractéristique majeure du photovieillissement est l’élastose solaire, une accumulation dans le derme réticulaire de fibres élastiques anormales, épaisses, associées à des protéines comme la fibronectine ou la fibrilline (Chen et al., 1986).

Le réseau de fibres de collagène , la protéine majoritaire de la MEC, est également désorganisé et le contenu total du derme en collagène est diminué (Talwar et al., 1995), en raison notamment d’une inhibition de la synthèse des pro‐collagènes I et III (Varani et al., 2001). Les métalloprotéinases matricielles (MMPs), des enzymes responsables de la dégradation des protéines de la MEC, jouent également un rôle majeur dans la désorganisation des collagènes observée dans la peau photoâgée (Quan et al., 2009).
IV.2. Les MMPs et leur rôle dans le photovieillissement
IV.2.a. Description des MMPs
Les MMPs sont des enzymes protéolytiques sécrétées ou membranaires qui régulent un grand nombre de processus biologiques par clivage de diverses protéines circulantes, membranaires ou extracellulaires. Elles interviennent notamment dans le remodelage tissulaire lors de la cicatrisation , la signalisation inter‐cellulaire, l’inflammation et l’angiogénèse (pour revue voir : Egeblad and Werb, 2002 ; Kerkela and Saarialho‐Kere, 2003 ; Sternlicht and Werb, 2001).
Les MMPs appartiennent à la superfamille des « metzincines » qui est caractérisée par la présence d’un motif très conservé de fixation du zinc au niveau du site catalytique. La famille des MMPs comprend 22 membres qui peuvent être divisés en 6 sous‐groupes : (1) les collagénases ; (2) les gélatinases ; (3) les stromélysines ; (4) les matrilysines ; (5) les MMPs associées à la membrane (membrane‐type, MT‐MMP) et (6) les autres MMPs. A elles toutes, les MMPs peuvent dégrader tous les composants de la MEC (tableau ci-dessous).
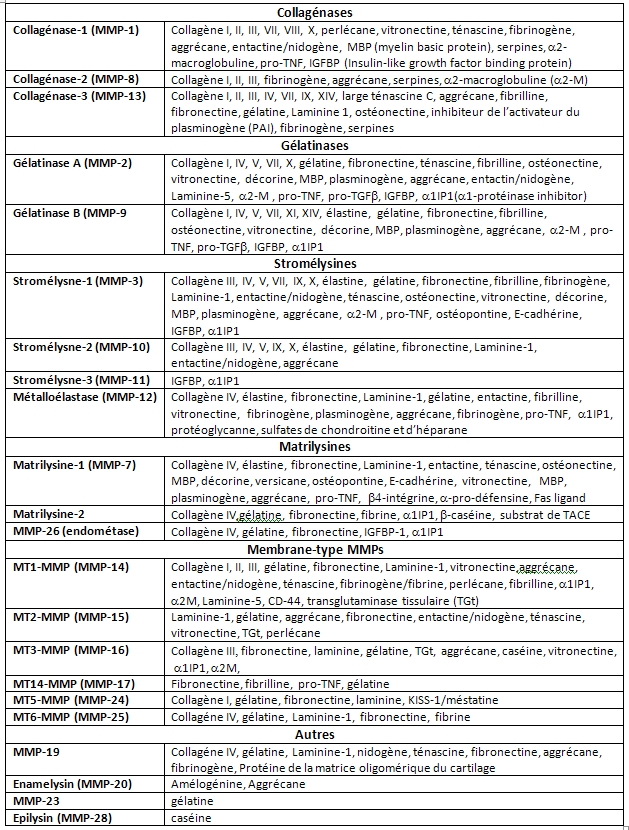
- Les substrats des MMPs
Les membres de la famille des MMPs diffèrent d’un point de vue structurel, ce qui explique la capacité de chacune des enzymes à dégrader un certain type de substrats (figure ci-dessous).
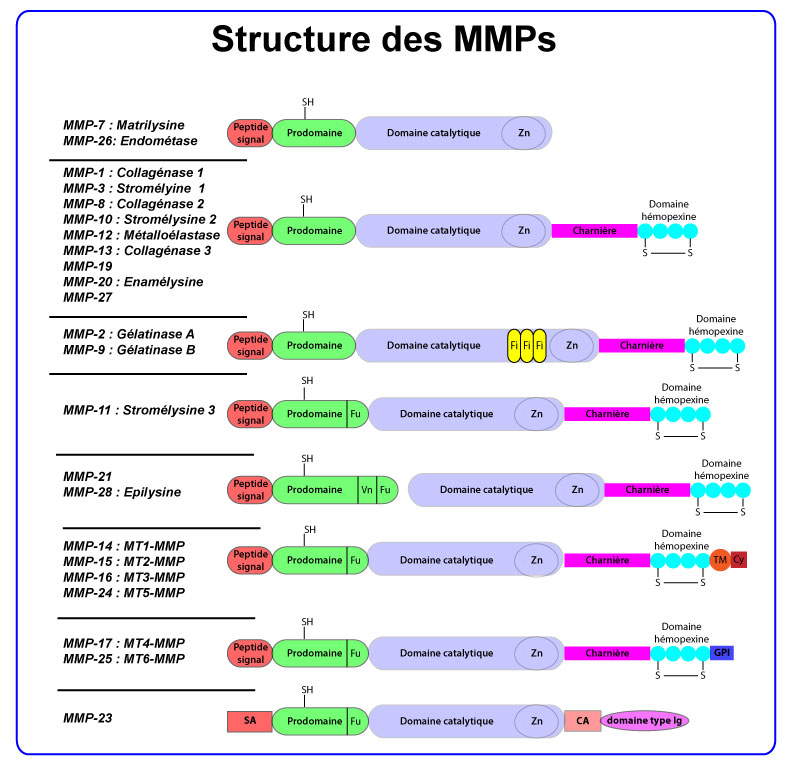
- Structure des MMPs
D’un point de vue structurel, les MMPs peuvent être divisées en huit groupes : les cinq premiers groupes rassemblent les MMPs sécrétées, les trois derniers correspondent aux MMPs transmembranaires (MTMMPs et CA‐MMP). La séquence « Pré » dirige les MMPs vers le réticulum endoplasmique au cours de la traduction. Le propeptide (Pro), qui contient un groupement thiol (SH) capable d’interagir avec le zinc, maintient les MMPs sous une forme inactive (zymogène). Le domaine catalytique contient un site de liaison au zinc (Zn). Le domaine hémopexine est connecté au domaine catalytique par une charnière polypeptidique (H). Il joue un rôle dans les interactions de l’enzyme avec les inhibiteurs des MMPs (TIMPs ), les molécules membranaires et les substrats protéiques. La première et la dernière répétition du domaine hémopexine sont reliées par un pont disulfure (S‐S). Les MMPs de type gélatinase possèdent des domaines ressemblant aux répétitions liant le collagène décrites sur la fibronectine (Fi). Le motif Fu permet la reconnaissance et l’activation de certaines MMPs sécrétées par les protéases de type furine. Les MT‐MMPs possèdent (1) un domaine transmembranaire (TM) et une courte région cytoplasmique (Cy) ou (2) un domaine d’ancrage au glycosylphosphatidylinositol (GPI). La MMP‐23 possède un signal d’ancrage N‐terminal (SA) qui la dirige vers la membrane cellulaire, ainsi qu’un domaine riche en cystéines (CA) et un domaine de type immunoglobuline (Ig).
Toutes les MMPs possèdent un signal peptidique N‐terminal (ou domaine « pré ») qui dirige la sécrétion de ces enzymes dans le réticulum endoplasmique (où le domaine « pré » est clivé) puis hors de la cellule. Ainsi, la plupart des MMPs sont constitutivement sécrétées après leur synthèse, sauf les MT‐MMPs qui possèdent un domaine transmembranaire et sont exprimées à la surface de la cellule. Le domaine « pré » est suivi d’un domaine « pro » qui maintient l’enzyme dans un état latent jusqu’à son clivage et l’activation conséquente de l’enzyme. Après le domaine « pro » se trouve le site catalytique de l’enzyme qui contient le domaine de liaison au zinc. A l’exception des MMP7, 26 et 23, toutes les MMPs possèdent un domaine homologue à l’hémopéxine connecté au site catalytique par une liaison peptidique. Ce domaine modifie l’affinité de l’enzyme pour certains substrats, influe sur son activité protéolytique et régule également la liaison des TIMPs (Tissue Inhibitors of MetalloProteinases), les inhibiteurs extracellulaires des MMPs.
IV.2.b. Régulation post‐traductionnelle des MMPs
Comme d’autres enzymes protéolytiques, les MMPs sont tout d’abord synthétisées et sécrétées comme des proenzymes (ou zymogènes). Leur activation extracellulaire est initiée par des MMPs préalablement activées ou par des protéases à spécificité sérine qui clivent le lien peptidique retenant le domaine « pro » (Chakraborti et al., 2003 ; Nagase and Woessner, 1999) (voir tableau ci-dessous).
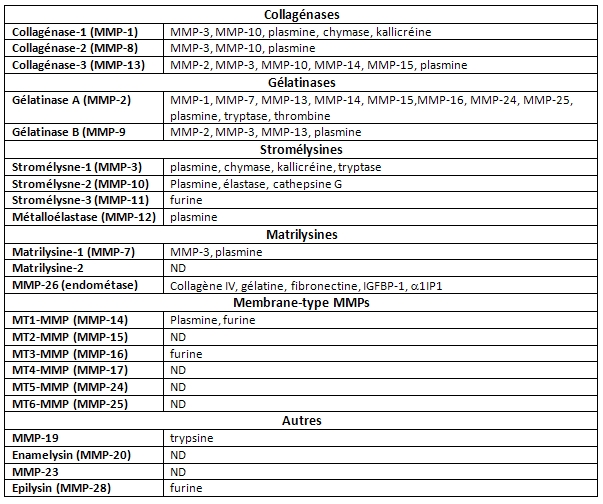
- Les activateurs des MMPs
Une dégradation contrôlée et coordonnée des composants de la MEC est nécessaire à la régulation de nombreux processus biologiques et l’activité des MMPs est donc contrôlée finement par leurs inhibiteurs, les TIMPs.
Les TIMPs forment une famille de 4 protéines sécrétées (TIMP 1 – 4) qui inhibent de façon réversible les MMPs selon un processus stoechiométrique (1 :1). Les TIMPs exercent leur activité inhibitrice par leur domaine N‐terminal qui se lie au site actif des MMPs et empêche la liaison de celles‐ci sur leurs substrats (Willenbrock and Murphy, 1994). Un déséquilibre de la synthèse des MMPs et des TIMPs, résultant en un excès de MMPs, joue un rôle déterminant dans de nombreux processus pathologiques et notamment dans la promotion de l’invasion tumorale (Westermarck and Kahari, 1999).
IV.2.c. MMPs, UV et photovieillissement
Les rayonnements UVB et SSL induisent l’expression de la MMP‐1 (collagénase interstitielle), de la MMP‐3 (stromélysine 1) et de la MMP‐9 (gélatinase 92kDa) dans la peau humaine in vivo (Fisher et al., 1996 ; Fisher et al., 1997 ; Quan et al., 2009). Cette induction est liée à la rapide activation des MAPK puis des facteurs AP‐1 qui régulent l’activité du promoteur des MMP‐1, ‐3 et ‐9 (Brenneisen et al., 2002 ; Fisher and Voorhees, 1998). L’inhibiteur TIMP‐1 est également induit par les UV parallèlement à l’induction des MMPs (Fisher et al., 1997). Les études menées in vitro sur des cultures de fibroblastes et des peaux reconstruites ont permis de montrer que les fibroblastes du derme étaient les producteurs majoritaires des MMPs en réponse à l’exposition UVB et SSL, suite à
une activation paracrine par les kératinocytes (Fagot et al., 2002 ; Fagot et al., 2004). Toutefois, une étude récente montre que, dans la peau in vivo, les MMPs seraient induites majoritairement dans
l’épiderme après une irradiaiton SSL (Quan et al., 2009), suggérant une régulation paracrine du derme vers l’épiderme. Les deux compartiments cutanés seraient donc impliqués dans la régulation de la production et de la sécrétion de MMPs.
Les UVA induisent également la sécrétion de MMP‐1 dans les fibroblastes en culture, dans les peaux reconstruites et dans la peau in vivo (Bernerd and Asselineau, 1998 ; Scharffetter et al., 1991). Cette induction est liée à l’activation du facteur AP‐1 par les ROS (Wenk et al., 1999). En raison de leurs propriétés de pénétration dans le derme, les UVA sont des acteurs majeurs dans les mécanismes menant aux altérations dermiques associées au photovieillissement (Bernerd and Asselineau, 1998).
Ensemble, les MMP‐1, ‐3 et ‐9 possèdent la capacité de dégrader la plupart des composants du derme. La MMP‐1 initie le clivage des collagènes fibrillaires de type I et III en coupant chaque fibre de la triple hélice constituant le collagène. Une fois clivé par la MMP‐1, le collagène peut être fragmenté par les MMP‐3 et ‐9. Les MMPs induites par les UV joueraient donc un rôle déterminant dans le processus de photovieillissement en participant à la dégradation et à la désorganisation de la MEC.
V. Photocarcinogénèse
Les altérations moléculaires, cellulaires et tissulaires induites par les UV, que nous avons évoquées tout au long de ce chapitre, contribuent, à long terme, au processus complexe menant au développement des carcinomes cutanés (figure 28).
Le gène suppresseur de tumeurs p53 est souvent qualifié de « gardien du génome » en raison de son rôle dans le contrôle du cycle cellulaire et de l’apoptose pour répondre à la présence de dommages dans l’ADN. Il est l’un des acteurs majeurs du processus de carcinogénèse. Le développement rapide de carcinomes cutanés chez les souris déficientes pour p53 (p53 ‐/‐) illustre l’importance de ce gène dans le contrôle de la promotion tumorale (Li et al., 1998b). Chez l’homme, de nombreuses études ont identifié la présence de mutations affectant le gène p53 dans les carcinomes cutanés : des mutations de p53 sont en effet présentes dans 41 à 90% des CSC (Bolshakov et al., 2003 ; Brash et al., 1991 ; Nelson et al., 1994 ; Ziegler et al., 1994) et dans 33 à 56% des CBC (Bolshakov et al., 2003 ; Kim et al., 2002 ; Rady et al., 1992 ; Soehnge et al., 1997 ; Zhang et al., 2001 ; Ziegler et al., 1993). La répartition de ces mutations n’est pas aléatoire : certains « points chauds » particulièrement susceptibles de porter une mutation provoquant l’inactivation fonctionnelle de p53 ont été décrits (Brash et al., 1991 ; Ziegler et al., 1993). Plusieurs de ces sites sont des séquences CpG, fréquemment méthylées. La formation préférentielle des CPD au niveau des séquences contenant une 5‐mC et le caractère hautement mutagène de ce type de séquence pourrait permettre d’expliquer la présence de ces points chauds (Drouin and Therrien, 1997 ; You et al., 2000). La majorité des mutations touchant le gène p53 sont des transversions C→T et/ou CC→TT, qui constituent la signature spécifique des UV.
La formation de mutations dans le gène p53 serait un évènement précoce du développement des cancers de la peau. En effet, des mutations spécifiques des UVB ont été identifiées sur le gène p53 dans 75 à 80% des KA, les précurseurs des CSC (Agar et al., 2004 ; Ziegler et al., 1994). De plus, de nombreux clones de kératinocytes portant des mutations sur le gène p53 sont présents dans la peau normale exposée au soleil (Jonason et al., 1996 ; Nakazawa et al., 1994). Ces clones se développent à partir de la JDE et/ou des follicules pileux et sont d’autant plus fréquents et larges que l’exposition solaire est chronique. Outre leur rôle dans l’initiation de la tumorigénèse, les UV pourraient donc également promouvoir le développement tumoral en induisant l’apoptose des cellules endommagées contenant une protéine p53 fonctionnelle (contrôle cellulaire ou « cellular
proofreading ») et en favorisant l’expansion clonale des cellules déficientes en p53.
Selon les modèles hypothétiques décrivant le processus multi‐étapes de la carcinogénèse, l’altération de deux gènes « gardiens « (« gatekeeper genes ») pourrait mener au cancer (Kinzler and Vogelstein, 1997). Dans les carcinomes cutanés, l’un de ces évènements impliquerait probablement p53 et la mutation d’un autre gène (impliqué par exemple dans le contrôle du cycle cellulaire ou la prolifération) mènerait à la transformation maligne. Suite à l’expansion clonale des kératinocytes présentant des altérations génétiques, l’émergence de la tumeur est finalement favorisée par l’immunosuppression induite par les UV qui permet aux cellules transformées d’échapper à la surveillance immunitaire anti‐tumorale. La forte incidence des CSC et de CBC dans les zones photoexposées chez les patients greffés et traités par des immunosuppresseurs illustre l’impact de l’immunosuppression sur le développement des carcinomes cutanés (Glover et al., 1997). Le rayonnement solaire peut donc être considéré comme un « carcinogène total » puisqu’il est capable à lui seul d’initier et de promouvoir le développement des cancers cutanés.
Bibliographie
Brash, D.E., A. Ziegler, A.S. Jonason, J.A. Simon, S. Kunala, and D.J. Leffell. 1996. Sunlight and sunburn in human skin cancer : p53, apoptosis, and tumor promotion. J Investig Dermatol Symp Proc. 1:136‐42.
Brenneisen, P., H. Sies, and K. Scharffetter‐Kochanek. 2002. Ultraviolet‐B irradiation and matrix metalloproteinases : from induction via signaling to initial events. Ann N Y Acad Sci. 973:31‐43.
Chakraborti, S., M. Mandal, S. Das, A. Mandal, and T. Chakraborti. 2003. Regulation of matrix metalloproteinases : an overview. Mol Cell Biochem. 253:269‐85.
de Gruijl FR. 2008. UV-induced immunosuppression in the balance. Photochem Photobiol. 84(1):2-9.
Del Bino, S., J. Sok, E. Bessac, and F. Bernerd. 2006. Relationship between skin response to ultraviolet exposure and skin color type. Pigment Cell Res. 19:606‐14.
Drouin, R., and J.P. Therrien. 1997. UVB‐induced cyclobutane pyrimidine dimer frequency correlates with skin cancer mutational hotspots in p53. Photochem Photobiol. 66:719‐26
Egeblad, M., and Z. Werb. 2002. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2:161‐74.
Fagot, D., D. Asselineau, and F. Bernerd. 2002. Direct role of human dermal fibroblasts and indirect participation of epidermal keratinocytes in MMP‐1 production after UV‐B irradiation. Arch Dermatol Res. 293:576‐83.
Fagot, D., D. Asselineau, and F. Bernerd. 2004. Matrix metalloproteinase‐1 production observed after solar‐simulated radiation exposure is assumed by dermal fibroblasts but involves a paracrine activation through epidermal keratinocytes. Photochem Photobiol. 79:499‐505.
Fisher, G.J., S.C. Datta, H.S. Talwar, Z.Q. Wang, J. Varani, S. Kang, and J.J. Voorhees. 1996. Molecularbasis of sun‐induced premature skin ageing and retinoid antagonism. Nature. 379:335‐9.
Fisher, G.J., Z.Q. Wang, S.C. Datta, J. Varani, S. Kang, and J.J. Voorhees. 1997. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 337:1419‐28.
Gilchrest, B.A., H.Y. Park, M.S. Eller, and M. Yaar. 1996. Mechanisms of ultraviolet light‐induced pigmentation. Photochem Photobiol. 63:1‐10.
Glover, M.T., J.J. Deeks, M.J. Raftery, J. Cunningham, and I.M. Leigh. 1997. Immunosuppression and risk of non‐melanoma skin cancer in renal transplant recipients. Lancet. 349:398.
Kerkela, E., and U. Saarialho‐Kere. 2003. Matrix metalloproteinases in tumor progression : focus on basal and squamous cell skin cancer. Exp Dermatol. 12:109‐25.
Kim, M.Y., H.J. Park, S.C. Baek, D.G. Byun, and D. Houh. 2002. Mutations of the p53 and PTCH gene in
basal cell carcinomas : UV mutation signature and strand bias. J Dermatol Sci. 29:1‐9.
Kinzler, K.W., and B. Vogelstein. 1997. Cancer‐susceptibility genes. Gatekeepers and caretakers. Nature. 386:761, 763.
Kripke, M.L. 1974. Antigenicity of murine skin tumors induced by ultraviolet light. J Natl Cancer Inst. 53:1333‐6.
Kripke, M.L., and M.S. Fisher. 1976. Immunologic responses of the autochthonous host against tumors induced by ultraviolet light. Adv Exp Med Biol. 66:445‐9.
Kripke, M.L. 1984. Immunological unresponsiveness induced by ultraviolet radiation. Immunol Rev. 80:87‐102.
[Maeda, K., and M. Hatao. 2004. Involvement of photooxidation of melanogenic precursors in prolonged pigmentation induced by ultraviolet A. J Invest Dermatol. 122:503‐9.-<http://www.nature.com/jid/journal/v...
[Nagase,]>
http://www.jbc.org/content/274/31/2...]
Nelson, M.A., J.G. Einspahr, D.S. Alberts, C.A. Balfour, J.A. Wymer, K.L. Welch, S.J. Salasche, J.L. Bangert, T.M. Grogan, and P.O. Bozzo. 1994. Analysis of the p53 gene in human precancerous actinic keratosis lesions and squamous cell cancers. Cancer Lett. 85:23‐9.
Norval, M. 2006. The mechanisms and consequences of ultraviolet‐induced immunosuppression. Prog Biophys Mol Biol. 92:108‐18.
[Rady, P., F. Scinicariello, R.F. Wagner, Jr., and S.K. Tyring. 1992. p53 mutations in basal cell carcinomas. Cancer Res. 52:3804‐6._>http://cancerres.aacrjournals.org/c...]
Schwarz T. Mechanisms of UV-induced immunosuppression. Keio J Med. 2005 Dec ;54(4):165-71. Review.
Soehnge, H., A. Ouhtit, and O.N. Ananthaswamy. 1997. Mechanisms of induction of skin cancer by UV radiation. Front Biosci. 2:d538‐51.
Westermarck, J., and V.M. Kahari. 1999. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 13:781‐92.
Willenbrock, F., and G. Murphy. 1994. Structure‐function relationships in the tissue inhibitors of metalloproteinases. Am J Respir Crit Care Med. 150:S165‐70.
Ziegler, A., A.S. Jonason, D.J. Leffell, J.A. Simon, H.W. Sharma, J. Kimmelman, L. Remington, T. Jacks, and D.E. Brash. 1994. Sunburn and p53 in the onset of skin cancer. Nature. 372:773‐6.
Site réalisé avec SPIP 3.0.17 + AHUNTSIC

Visiteurs connectés : 57